|
| Temas de FC |
J.M. Marugán de Miguelsanz*,***, M.C. Torres Hinojal**, C. Alonso Vicente***
*Profesor Titular de Pediatría. Facultad de Medicina. Universidad de Valladolid. **Pediatra de Atención Primaria. Centro de Salud Huerta del Rey. Valladolid. ***Unidad de Gastroenterología pediátrica. Hospital Clínico Universitario de Valladolid
| Resumen
El hígado es un órgano central en la economía humana y cuenta con múltiples funciones. Las alteraciones de las pruebas de función hepática son la mayor parte de las veces inespecíficas, y su correcta interpretación constituye un reto diagnóstico para el pediatra. Las causas que pueden alterar las pruebas hepáticas son numerosas: primariamente, específicas del hígado, extrahepáticas o sistémicas con repercusión sobre el mismo, o bien ajenas al hígado (p. ej., miopatías). Debemos descartar una hiperbilirrubinemia conjugada ante toda ictericia prolongada más allá de las dos semanas de vida. Las transaminasas son los marcadores más sensibles de lesión hepatocelular, y el diagnóstico de una hipertransaminasemia mantenida requiere una actitud diagnóstica escalonada. Una elevación de las mismas puede ser inespecífica, o bien sugerente de hepatopatía aguda o crónica, y pueden ser normales o solo levemente elevadas, incluso en hepatopatías graves. Los marcadores más específicos de colestasis son la gamma-glutamiltranspeptidasa y la 5’nucleotidasa, mientras que los de síntesis hepática son la albúmina y el tiempo de protrombina. La ecografía es una excelente técnica diagnóstica en patología hepatobiliar, y la elastografía por ondas de choque ha supuesto un gran avance en el estudio de la fibrosis hepática. Finalmente, la biopsia hepática tiene todavía muchas indicaciones en la hepatopatía crónica, especialmente si no hay una causa clara, para confirmar ciertos diagnósticos, ante una posible enfermedad metabólica y, en caso de cronicidad, para estudiar sus consecuencias (fibrosis, cirrosis hepática). |
| Abstract
The liver is a central organ in the human anatomy and has multiple functions. Alterations in liver function tests are mostly non-specific, and their correct interpretation constitutes a diagnostic challenge for the pediatrician. The causes that can alter liver function tests are numerous: primarily, liver-specific, extrahepatic or systemic with an impact on the liver, or foreign to the liver (eg. myopathies). In any prolonged jaundice beyond two weeks of life, conjugated hyperbilirubinemia must be ruled out. Transaminases are the most sensitive markers of hepatocellular injury, and the diagnosis of mantained hypertransaminasemia requires a staggered diagnostic approach. |
Palabras clave: Hepatopatía en niños; Diagnóstico; Hipertransaminasemia.
Key words: Hepatopathy in children; Diagnosis; Hypertransaminasemia.
Pediatr Integral 2020; XXIV(1): 6 – 14
Aproximación diagnóstica al paciente con enfermedad hepática
Introducción
El hígado tiene numerosas funciones y las causas de hepatopatía o de alteración de las pruebas de función hepáticas son innumerables.
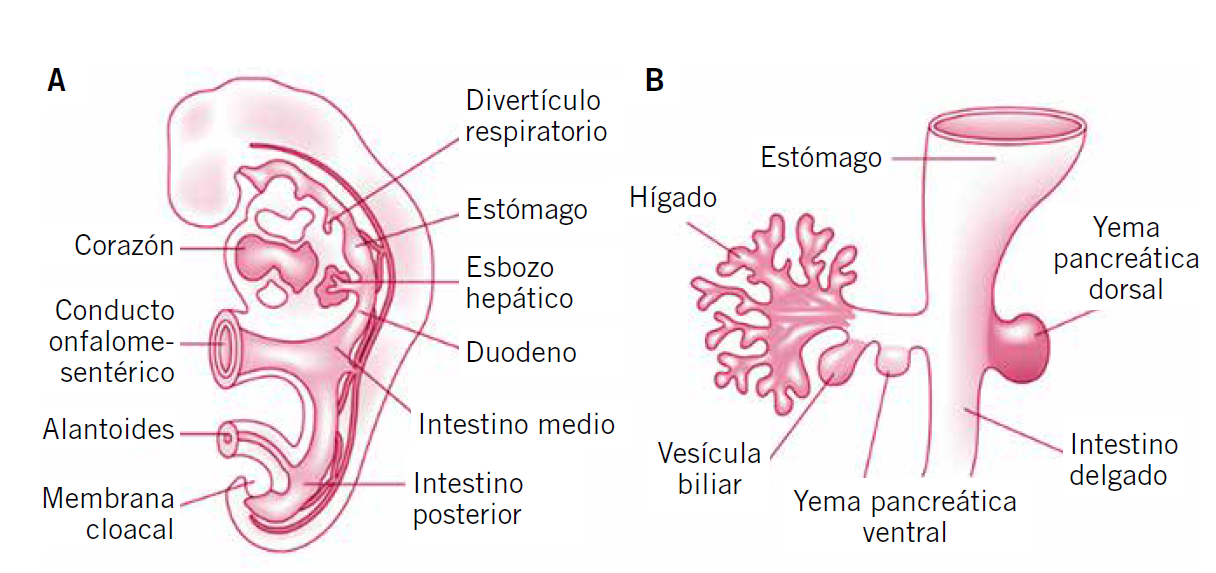
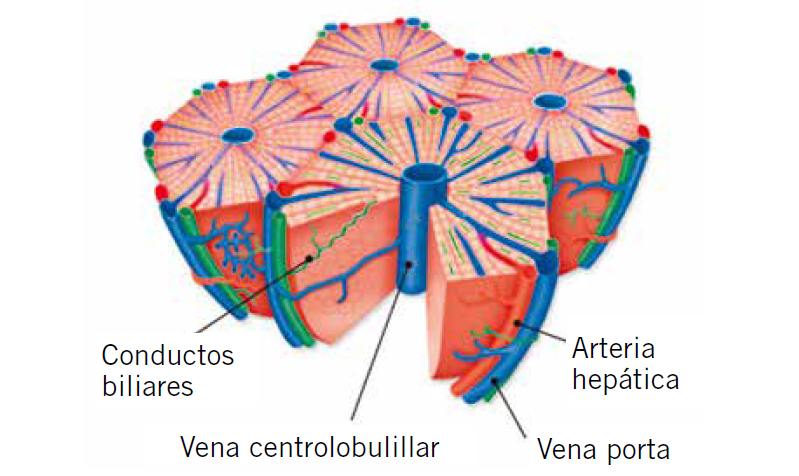
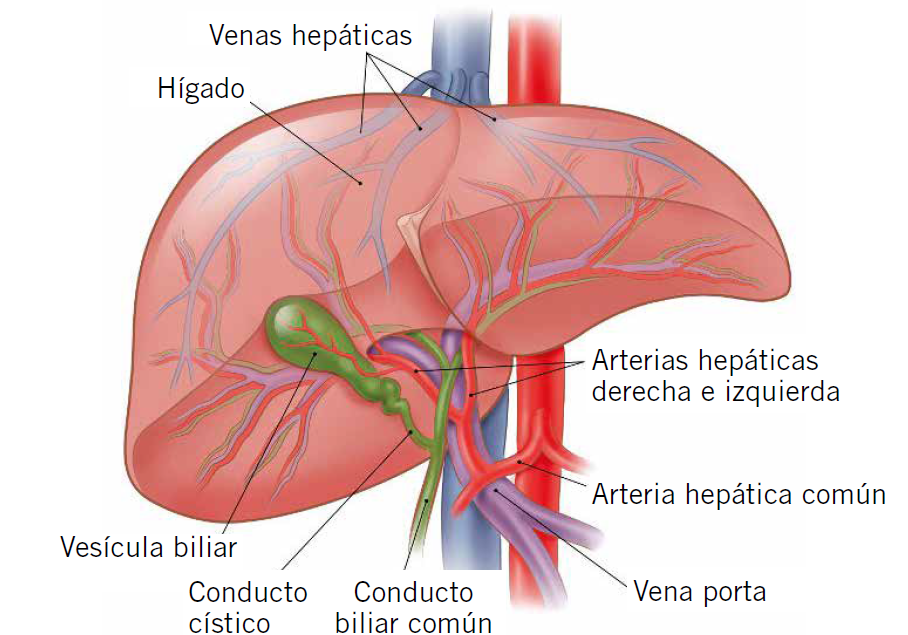
El hígado es un órgano central en la fisiología humana, especialmente en el metabolismo intermediario, e interacciona con otros muchos órganos y sistemas. Es la víscera de mayor tamaño de nuestra economía y tiene múltiples y complejas funciones. Entre otras, el hígado se encarga de la detoxificación, conversión de amonio en urea, almacenamiento de glucógeno y liberación de glucosa, síntesis de ácidos grasos y cetogénesis, síntesis proteica, de lipoproteínas, colesterol y fosfolípidos, de factores inmunitarios, de factores de la coagulación, depuración y eliminación de bilirrubina, y formación y secreción de bilis(1).
Todos los antígenos absorbidos por el tubo digestivo, microorganismos y otros factores sistémicos, así como la metabolización de fármacos y productos tóxicos, pasan necesariamente por el hígado, y por el efecto de todos ellos puede producirse enfermedad y alteración de las pruebas de función hepática.
El estudio de una posible enfermedad hepática es, por lo tanto, complejo, muy amplio y puede implicar a un gran número de enfermedades en el niño, aunque trataremos de simplificarlo a los aspectos fundamentales del mismo, aun a riesgo de poder olvidar hechos importantes y también relacionados. En el estudio de un paciente con enfermedad hepática, deberemos atender a las siguientes etapas del proceso diagnóstico.
Historia clínica
La historia clínica debe darnos la clave inicial del diagnóstico de cualquier hepatopatía, aunque la semiología es con frecuencia inespecífica. Los síntomas más conocidos y específicos de enfermedad hepática son: hepatomegalia, ictericia, acolia y coluria.
Una detallada anamnesis y exploración física son, como siempre, imprescindibles en la aproximación diagnóstica a las hepatopatías. Los antecedentes familiares, perinatales (infecciones, prematuridad, hipoxia intraparto), crecimiento y desarrollo, patologías asociadas, consumo de sustancias o fármacos, o síntomas sistémicos, deben recogerse y pueden orientar sobre la etiología del proceso.
Los síntomas y signos iniciales de hepatopatía aguda son con frecuencia inespecíficos, como por ejemplo, el típico cuadro prodrómico en las hepatitis agudas virales, con: astenia, intensa anorexia, náuseas, vómitos o dolor epigástrico. El dolor abdominal de causa hepática es infrecuente por incremento del tamaño del hígado, y cuando es más intenso, debe poner sobre la pista de patología y obstrucción de la vía biliar. En formas crónicas, el cuadro clínico puede ser aún más insidioso, inespecífico o incluso asintomático, y con frecuencia existe poca correlación entre la intensidad de los síntomas y la gravedad de la hepatopatía.
Al margen de otros síntomas de enfermedades sistémicas que analizaremos más adelante, los síntomas más específicos y conocidos de enfermedad hepática son: hepatomegalia, ictericia, acolia y coluria(2).
• Hepatomegalia. El hígado puede aumentar de tamaño por muchos mecanismos y la hepatomegalia puede ser debida a una enfermedad hepática o bien a una enfermedad generalizada. Los principales mecanismos de hepatomegalia son: inflamación, depósito, infiltración, congestión vascular u obstrucción biliar(3).
• Ictericia. Sugerente, aunque no siempre debida a hepatopatía. No vamos a repasar las numerosas causas de hiperbilirrubinemia neonatal, pero hay que recordar que ante una ictericia prolongada más allá de las dos semanas de vida, es obligatorio conocer la fracción conjugada de la bilirrubina (BR).
• Coluria. Cuando el contenido en pigmentos biliares en la orina es excesivo, como consecuencia, al igual que la acolia, de obstrucción biliar o lesión hepatocelular.
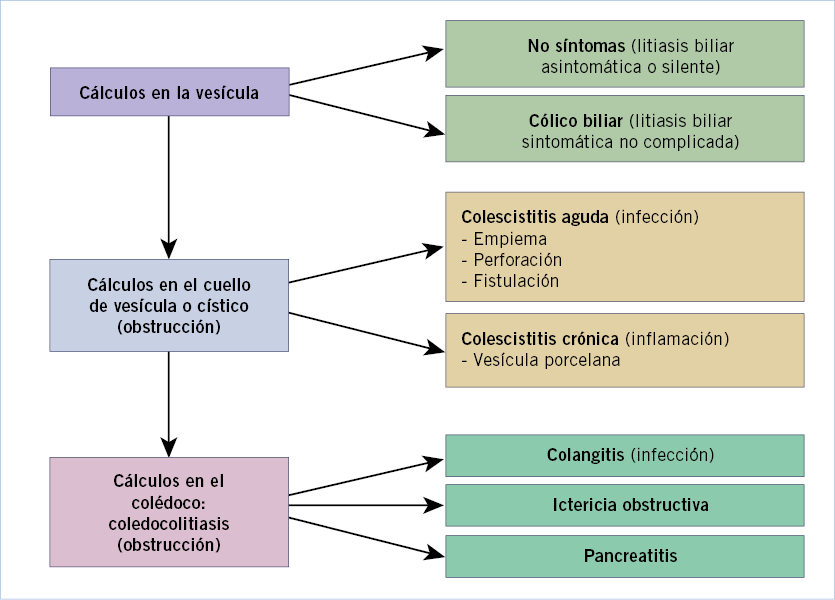
• Acolia. Cuando no hay un drenaje biliar normal al intestino. Se relaciona con las ictericias obstructivas, congénitas o adquiridas, o con las hepatitis agudas. Se debe sospechar una hepatopatía colestásica en un lactante con ictericia que presenta orinas colúricas o heces acólicas, y estudiar de forma inmediata los niveles de BR conjugada y total, ante la posibilidad de una atresia de vía biliar extrahepática(4,5).
• Otros síntomas sugerentes de hepatopatía pueden ser: prurito intenso y generalizado, sobre todo, en las hepatopatías que cursan con colestasis (aunque no guarda relación con el grado de hiperbilirrubinemia) o síntomas como: telangiectasias, eritema palmar, trastornos endocrinos, malnutrición, ascitis, esplenomegalia y hemorragia gastrointestinal por varices esofágicas en caso de hipertensión portal, que pueden aparecer en hepatopatías crónicas más graves.
En el fallo hepático, cuando se afecta gravemente la función del hígado, puede observarse, además: déficit de vitaminas liposolubles si existe colestasis, hemorragias por déficit de vitamina K o disfunción hepática, o síntomas ya más tardíos de encefalopatía, debidos a la hiperamoniemia. Las colangitis de repetición pueden aparecer en hepatopatías colestásicas infantiles(5).
En un paciente en el que la elevación de transaminasas se detecta de forma casual, es raro el hallazgo de signos en la exploración física. No obstante, debemos buscar todos los referidos anteriormente. A título de ejemplo, ante colestasis y un fenotipo peculiar, deberemos pensar en el síndrome de Alagille, si existe deterioro neurológico podría orientar a una metabolopatía, o ante la sospecha de enfermedad de Wilson, deberemos buscar el característico anillo de Kayser-Fleischer en el examen oftalmológico corneal.
Pruebas de función hepática
Las alteraciones de las pruebas de función hepática son la mayor parte de las veces inespecíficas, y su correcta interpretación constituye un reto diagnóstico para el pediatra. Existen pruebas bioquímicas y pruebas funcionales hepáticas.
Ante la sospecha de hepatopatía, deberemos recurrir a las conocidas como pruebas de función hepática. Su alteración, especialmente una hipertransaminasemia leve a moderada, es también con frecuencia, un hallazgo inespecífico y casual en múltiples problemas, a veces, banales, o incluso asintomáticos, no primariamente hepáticos, y un reto diagnóstico frecuente en Atención Primaria.
En un estudio analítico en adolescentes estadounidenses entre 12 y 19 años, se observó aumento aislado de transaminasas en un 7,4% de los casos, siendo más frecuente en varones(6). Sin embargo, en múltiples hepatopatías, incluso potencialmente graves, sí es una manifestación frecuente. Puede considerarse, por tanto, un marcador sensible, pero poco específico de daño hepático.
No existe una prueba única que valore globalmente el hígado. Para aumentar su rendimiento diagnóstico, debemos tener en cuenta conjuntamente varias de ellas y, en todo caso, conocer su utilidad en unos u otros procesos para interpretarlas adecuadamente. Las denominadas pruebas de función hepática incluyen determinaciones bioquímicas y también pruebas dinámicas que valoran su capacidad funcional (sobre todo, metabólica y excretora)(1,7,8).
Las pruebas bioquímicas son poco sensibles, ya que pueden ser normales en hepatopatías graves, y sugieren solo un tipo de afectación hepática, bien lesión hepatocelular o bien colestasis, pero no informan de la etiología de la misma. También son poco específicas, pues pueden alterarse en multitud de problemas extrahepáticos.
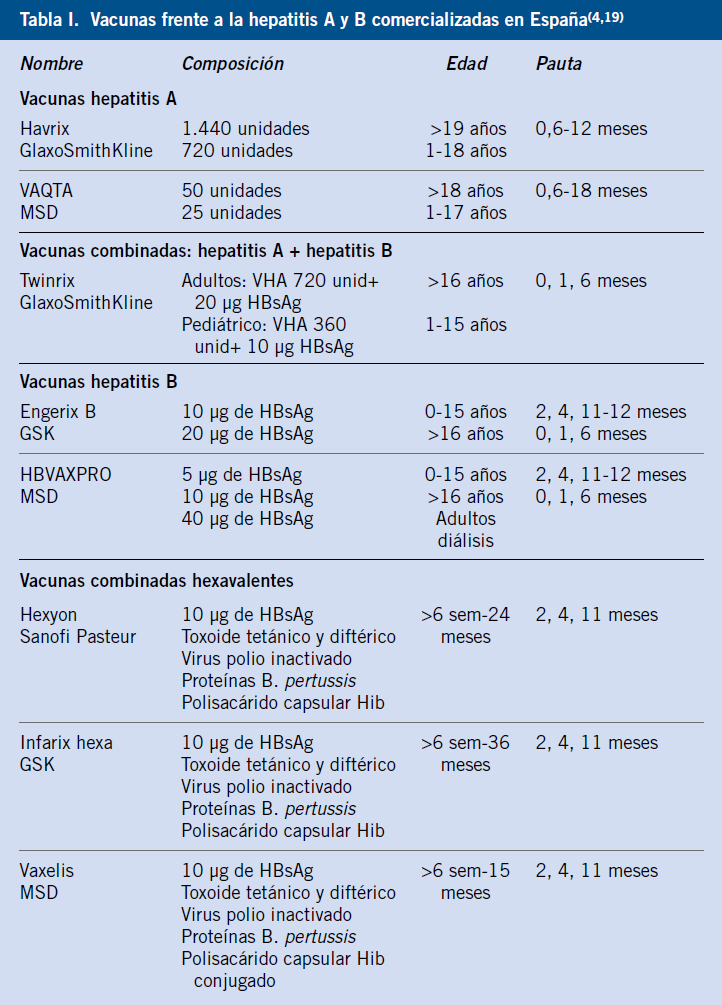
En dichas pruebas bioquímicas, el rango utilizado varía según el laboratorio y población de referencia, pero hasta un 2,5% de la población normal sin enfermedad hepática alguna, puede tener valores elevados de una prueba hepática concreta. Una prueba bioquímica anormal (preferiblemente, al menos, dos veces el valor superior de la normalidad) debe, por lo tanto, ser interpretado en el contexto clínico, por lo que la historia cuidadosa es clave en su interpretación(1). Enumeraremos, a continuación, las principales pruebas útiles para el diagnóstico de las enfermedades hepáticas (Tabla I).
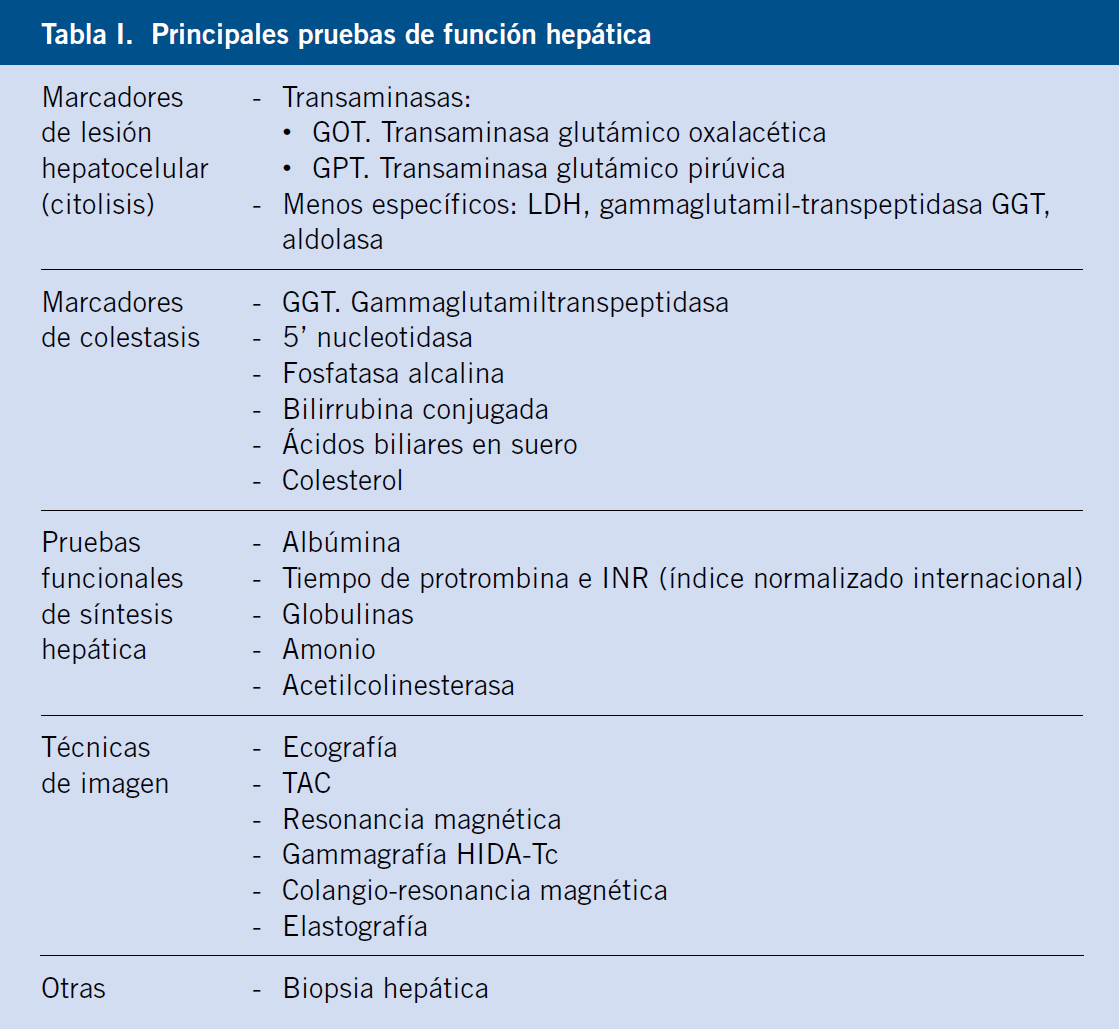
Marcadores de lesión hepatocelular (citolisis)(2,7,9)
Las transaminasas son los marcadores más sensibles de lesión hepatocelular. Una elevación de las mismas puede ser inespecífica, o bien sugerente de hepatopatía aguda o crónica, y pueden ser normales o solo levemente elevadas incluso en hepatopatías graves.
Transaminasas o aminotransferasas
En procesos inflamatorios agudos con lesión o necrosis del hepatocito, son los marcadores más sensibles de lesión hepática (hepatitis). La simple lesión de membrana ya permite la salida de enzimas, sin que se precise la existencia necesariamente de necrosis(1). Son dos:
• Transaminasa glutámico-oxalacética o aspartato-aminotransferasa (GOT, AST). No solo se encuentra en el hígado, sino también en: músculo esquelético, corazón, pulmón, páncreas, riñones, cerebro, hematíes y leucocitos. En el hepatocito, tiene una fracción citoplasmática y otra mitocondrial más abundante, por lo que su mayor elevación en hepatopatías agudas suele traducir una lesión hepatocelular más profunda y grave.
• Transaminasa glutámico-pirúvica o alanín-aminotransferasa (GPT, ALT). Es casi exclusiva del hígado, de localización citoplasmática, pudiendo traducir su elevación una lesión superficial del hepatocito. Se encuentra en menor medida en el músculo, y puede elevarse también en enfermedades de este tejido; de hecho, una miopatía o un ejercicio intenso pueden aumentar ambas transaminasas, en mayor proporción la GOT, en este caso de origen muscular.
Los niveles de ambas suelen correlacionarse poco con la clínica y carecen, en general, de valor pronóstico, aunque en la hepatitis fulminante, tras su ascenso inicial, pueden descender finalmente por depleción de hepatocitos.
Una hipertransaminasemia leve o moderada, inferior a 10 veces el límite superior de la normalidad, puede ser inespecífica o bien sugestiva de una lesión hepática crónica. También observaremos una elevación moderada de las mismas en la lesión hepática secundaria a un traumatismo abdominal cerrado, lo que permite un diagnóstico precoz del mismo(9).
Por el contrario, elevaciones graves de transaminasas, con valores superiores a 500 U/L (o por encima de 10 veces el límite de la normalidad), suelen indicar lesión hepatocelular primaria, tratándose, en la mayoría de los casos, de: hepatitis agudas víricas, necrosis por fármacos o tóxicos, exacerbación de una hepatitis autoinmune o hipoxia/hipoperfusión grave (shock)(7,10).
En la lesión vírica aguda, la GPT suele superar la cifra de GOT. Sin embargo, predomina la GOT sobre la GPT en elevaciones moderadas debidas a ingesta elevada de alcohol, en necrosis aguda fulminante en patología aguda, y en la cirrosis.
Una elevación casi exclusiva de GOT suele tener más un origen hemolítico, cardiaco o muscular, o puede tratarse de una macro-GOT.
Otros enzimas de citolisis
Pueden relacionarse también con lesión hepática, pero tienen menos rentabilidad diagnóstica que las transaminasas. Así, la LDH, que resulta poco específica, aunque su elevación mantenida, junto al de fosfatasa alcalina, se ha asociado, en ocasiones, a neoplasias hepáticas. También, la gammaglutamil-transpeptidasa (GGT), que veremos a continuación como enzima de colestasis, en menor medida, puede ser marcador de citolisis. Finalmente, la aldolasa (enzima procedente sobre todo del músculo) puede encontrarse algo elevada también en las hepatopatías agudas, a diferencia de la creatinquinasa (CK), exclusiva del tejido muscular. Ello puede tener utilidad para diferenciar una hipertransaminasemia debida a hepatopatía o miopatía.
Marcadores de colestasis
Los marcadores más específicos de colestasis son la gamma-glutamiltranspeptidasa y la 5’nucleotidasa, mientras que los de capacidad funcional o síntesis hepática, son la albúmina y el tiempo de protrombina.
En el patrón colestásico, ante una obstrucción o inflamación de la vía biliar, predomina la elevación de las siguientes enzimas y metabolitos, sobre el aumento de las transaminasas(2,5,7):
• Gamma-glutamil-transpeptidasa (GGT). Se localiza en el hepatocito (enzima microsomal ligado a membranas) y en el epitelio biliar (en mucha menor medida en otros órganos, pero no en el hueso, como sí ocurre con la fosfatasa alcalina). El alcohol y muchos fármacos pueden inducir su síntesis hepática. En los dos primeros meses de vida, pueden alcanzarse valores dobles o triples que en el niño mayor. Suele elevarse discretamente en hepatopatías agudas y también en tumores, pero especialmente en las colestasis, donde es el enzima más sensible, aunque por lo comentado antes, poco específico.
• -5’-nucleotidasa. Sensibilidad parecida a la fosfatasa alcalina en el diagnóstico de colestasis, pero más específica que ella, ya que no se encuentra en el hueso. Es de origen hepático predominante, aunque no exclusivo. Ya con menor especificidad que estas dos primeras, tenemos:
• Fosfatasa alcalina (ALP, FA). Es menos específica, debido a que su fracción predominante procede del hueso. En caso de duda, para diferenciar el origen de la elevación, puede recurrirse a la determinación de isoenzimas de la misma.
• Bilirrubina. Una elevación de la BR conjugada superior al 20% de la cifra de bilirrubina total, indicará casi siempre una enfermedad hepática o de las vías biliares, y si es superior al 50%, sugerirá una obstrucción extrahepática(7). Solo la forma conjugada de BR aparece en la orina, con lo que una hiperbilirrubinuria implicará la existencia de enfermedad hepatobiliar.
• Ácidos biliares en suero. Su cifra dependerá: del flujo sanguíneo hepático, de su captación, de su secreción biliar, absorción intestinal y circulación enterohepática. Su elevación es una determinación sensible en la enfermedad hepatobiliar, pero también inespecífica, y no aporta ventajas sobre las pruebas clásicas ya comentadas.
• Colesterol. Suele estar aumentado en la colestasis, pero puede estar descendido en hepatopatías agudas graves, como la hepatitis aguda.
• Urobilinógeno en orina. Esta sustancia es producida en el intestino por acción de la flora intestinal a partir de la bilirrubina excretada en la bilis. Puede aumentar en las hemólisis importantes, hepatitis o cirrosis, y puede ser negativo en la obstrucción de las vías biliares.
Pruebas funcionales de síntesis hepática
Enumeramos, a continuación, las principales pruebas que valoran la capacidad de síntesis o funcional hepática, más útiles en nuestra práctica clínica, entre las que destacan la albúmina y el tiempo de protrombina(7).
• Albúmina. Producida exclusivamente en el hígado, puede ser un índice de su capacidad de síntesis, aunque tardío, por su larga vida media (20 días), precisándose varias semanas de importante disfunción hepática para observar su reducción. Sí puede ser un indicador pronóstico en pacientes con cirrosis hepática. Debemos recordar que puede descender, además en: malnutrición proteica, cambios en la volemia, síndrome nefrótico o enteropatía pierde-proteínas. La prealbúmina tiene una corta semivida (2 días), por lo que puede ser más sensible en cuadros agudos, aunque se afecta menos en la enfermedad hepática.
• Tiempo de protrombina o índice normalizado internacional (INR). En el hígado se sintetizan todos los factores de la coagulación, con la posible excepción del factor VIII. En lesión hepática grave, se observa descenso de la síntesis, sobre todo del factor V, y de los factores vitamina K dependientes (II, VII, IX y X), con alargamiento especialmente del tiempo de protrombina. Un descenso mantenido del factor VII es un indicador de mal pronóstico en hepatopatías fulminantes(5,7,11).
El tiempo de protrombina está prolongado (con frecuencia inferior al 50%) en la evolución a una hepatitis fulminante o en las hepatopatías crónicas con cirrosis, cuando hay un deterioro superior al 80% de la función hepática. Se producirán hemorragias, sobre todo, si además hay hipofibrinogenemia (factor I), típica también de la lesión hepática grave. Para ver si el tiempo de protrombina alargado (INR) puede ser debido solo a un déficit de vitamina K, se puede administrar esta por vía parenteral (5-10 mg), debiendo producirse su normalización en 12-24 horas o, al menos, un ascenso del 30% de dicho índice(5).
• Globulinas. Puede ser útil la determinación de sus fracciones, cuyos hallazgos no son típicos, pero pueden ayudar en el diagnóstico de la hepatopatía crónica. La fracción alfa-1 está muy disminuida en el déficit de alfa-1-antitripsina, la alfa-2 en la enfermedad de Wilson, y la fracción gamma se eleva en la hepatitis crónica autoinmune (niveles elevados de Ig G).
• Amoniaco. En estadios avanzados de disfunción hepática, no se produce la conversión del amoniaco tóxico en urea para su eliminación renal, presentando hiperamoniemia, con la consiguiente encefalopatía hepática y coma.
• La acetil-colinesterasa sérica no es influenciada por la hipoalbuminemia, y su descenso suele indicar también lesión hepática.
• Finalmente, la determinación de glucosa es poco específica, aunque la hipoglucemia es típica del fallo hepático agudo (por defecto en la glucogenolisis y neoglucogénesis).
• Pruebas dinámicas de función hepática. Otras pruebas, como al aclaramiento del verde de indocianina (especialmente útil en la cirugía hepática y en el trasplante), de aclaramiento de sustancias, o pruebas del aliento, no están al alcance de la mayor parte de centros(7).
Técnicas de imagen
La ecografía en manos expertas es una excelente técnica diagnóstica en patología hepato-biliar. La elastografía por ondas de choque ha supuesto un gran avance en el estudio de la fibrosis hepática, evitando muchas veces la biopsia tradicional.
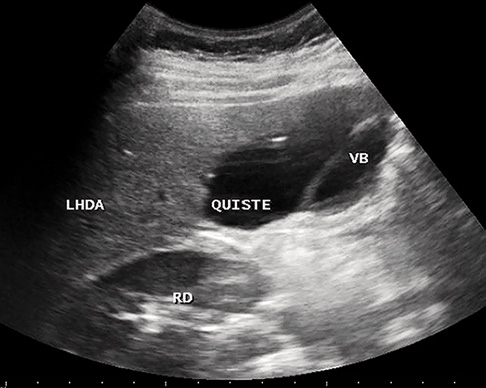
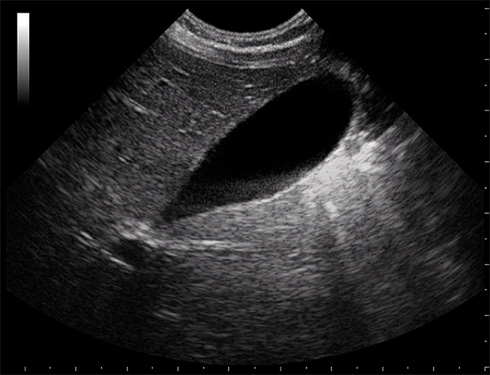
La gran utilidad de la ecografía ha desplazado, como primera técnica, a otras exploraciones clásicas, aunque en casos seleccionados puede precisarse otra técnica: radiológica, endoscópica, vascular con contraste, o de medicina nuclear, para concretar el diagnóstico. En una gammagrafía hepatobiliar (HIDA-Tc99m), tras varios días de inducción con fenobarbital, la eliminación de isótopo a intestino descarta la atresia biliar extrahepática (ABE). La ecografía permite con frecuencia sospechar un hígado graso no alcohólico (hiperecogenicidad difusa), ha desplazado a la colangiografía en el estudio de problemas biliares (litiasis, quistes) y permite detectar la presencia de ascitis. Una vesícula biliar pequeña o ausente, y la no visualización del colédoco pueden hacer pensar en una ABE. La eco-doppler permite sospechar una hipertensión portal. El TAC no aporta mucha más información sobre una ecografía en manos expertas, excepto para el estudio de lesiones focales (tumores, quistes o abscesos)(5), pero sí la resonancia magnética.
La elastografía hepática por ondas de choque (Fibroscan®) ha supuesto una gran ayuda en el seguimiento de pacientes con hepatopatía crónica y riesgo de fibrosis. Determina el grado de rigidez del hígado, y es un método inocuo que permite la detección temprana y el seguimiento de la fibrosis hepática. Su uso puede modificar el pronóstico de la misma y evitar el uso reiterado de biopsias hepáticas para su estudio y control evolutivo(12).
Biopsia hepática
La biopsia hepática tiene todavía muchas indicaciones en la hepatopatía crónica, especialmente si no hay una causa clara, para confirmar ciertos diagnósticos, ante una posible enfermedad metabólica y, en caso de cronicidad, para estudiar sus consecuencias (fibrosis, cirrosis hepática).
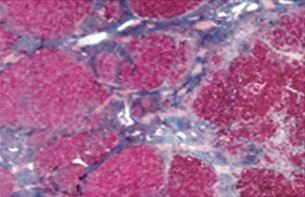
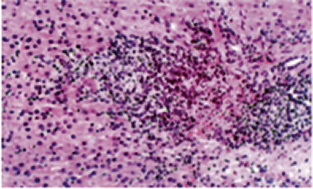
Crucial en el diagnóstico de una posible hepatopatía de etiología no aclarada o bien de causa conocida, pero de duración mayor de 6 meses (formas crónicas). Suele realizarse un estudio de microscopía óptica convencional y técnicas citoquímicas y, solo en casos concretos, de microscopía electrónica. En la muestra obtenida, también pueden realizarse análisis enzimáticos, ante la sospecha de una metabolopatía o enfermedad de depósito, demostrando el depósito anormal de sustancias (cobre, hierro o metabolitos específicos)(5).
Causas de alteración en las pruebas de función hepática en niños
Las causas que pueden alterar las pruebas hepáticas son numerosas: primariamente, específicas del hígado, extrahepáticas o sistémicas con repercusión sobre el mismo, o bien ajenas al hígado (p. ej., miopatías).
Describiremos muy brevemente, a continuación, las principales causas de alteración de las pruebas hepáticas (Tabla II), sin ánimo de ser exhaustivos.

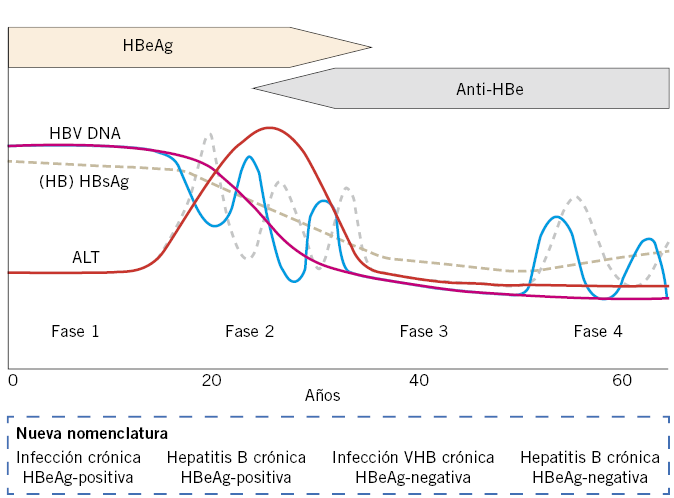
• Hepatitis viral. Aunque ha disminuido en gran medida su incidencia, los virus siguen siendo una de las causas más frecuentes de hepatitis. Se estudiará la serología de los virus hepatotropos mayores (VHA, B, C, D, E, G) y menores (citomegalovirus, virus de Epstein-Barr y herpes simple). Otros muchos virus causantes de enfermedad sistémica pueden afectar también al hígado, produciendo una hepatitis aguda de intensidad en general menor que la observada con los virus hepatotropos (rubeola, sarampión, varicela, parvovirus B19, echovirus y adenovirus, entre otros). Solo algunos virus hepatotropos mayores (VHB, VHC, VHD y VHG) pueden progresar a hepatitis crónica (elevación de transaminasas superior a 6 meses) y favorecer el desarrollo de cirrosis y hepatocarcinoma(9,13).
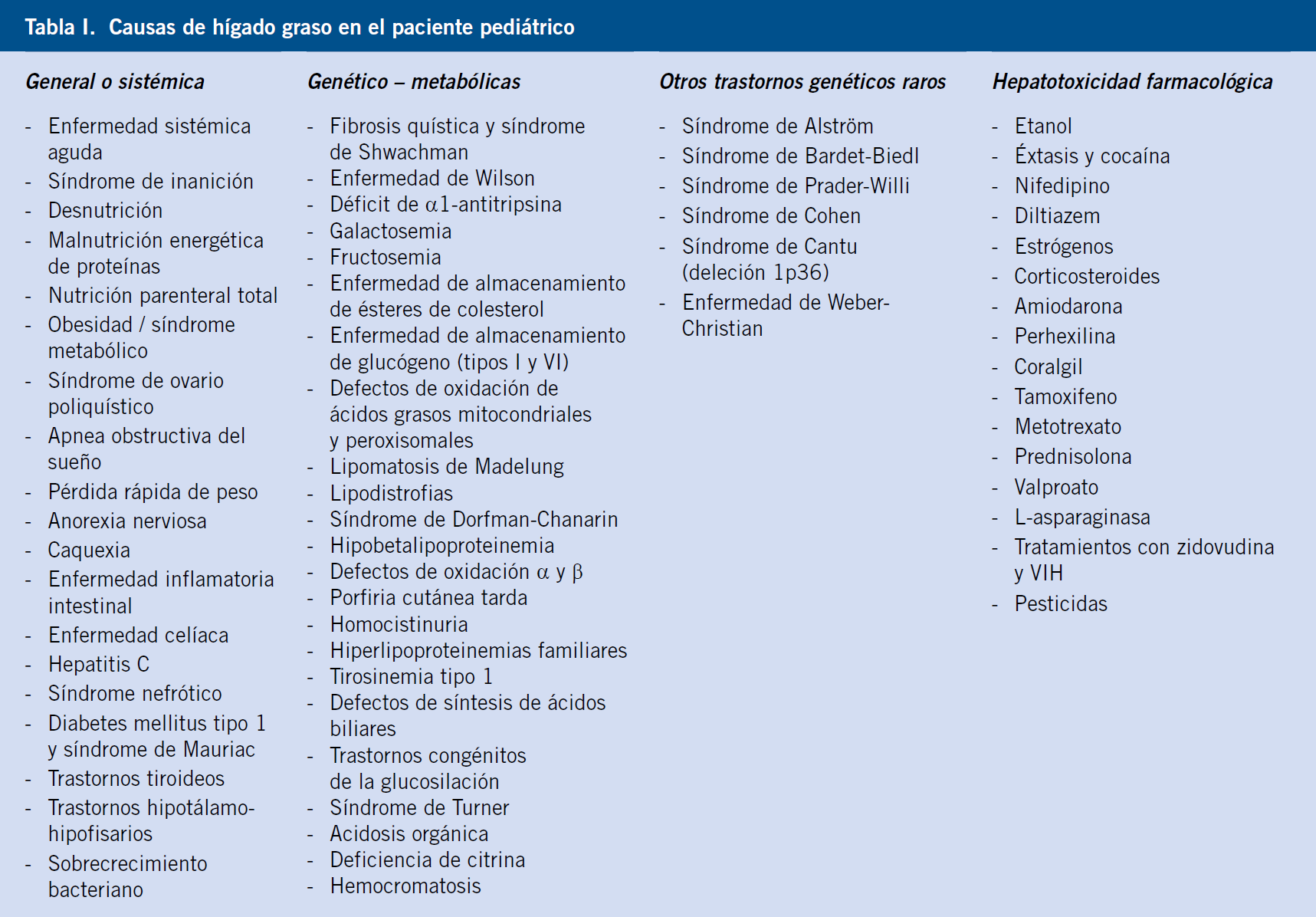
• Enfermedad hepática grasa no alcohólica (EHGNA). Se trata de la patología hepática más frecuente en edad pediátrica en el mundo civilizado. La obesidad es con mucho el principal factor etiológico del hígado graso, seguido en orden de frecuencia, por la diabetes mellitus y la hiperlipemia(14). Consiste en una esteatosis macrovesicular, con posible evolución a esteatohepatitis y fibrosis, probablemente mediada por una serie de factores genéticos y ambientales. En el proceso diagnóstico, la ecografía y la hipertransaminasemia constituyen los pilares del diagnóstico, constituyendo la biopsia hepática, el método de confirmación diagnóstica de la entidad, no siempre necesaria.
• Hepatitis por fármacos, tóxicos o productos de herboristería. Sus manifestaciones son muy variables, con frecuencia desde una hipertransaminasemia asintomática, hasta una insuficiencia hepática fulminante. La relación en el tiempo entre la ingesta del producto y el comienzo y la resolución de la lesión hepática son el aspecto más importante para el diagnóstico, ya que no cuenta con ningún hecho específico(13,15).
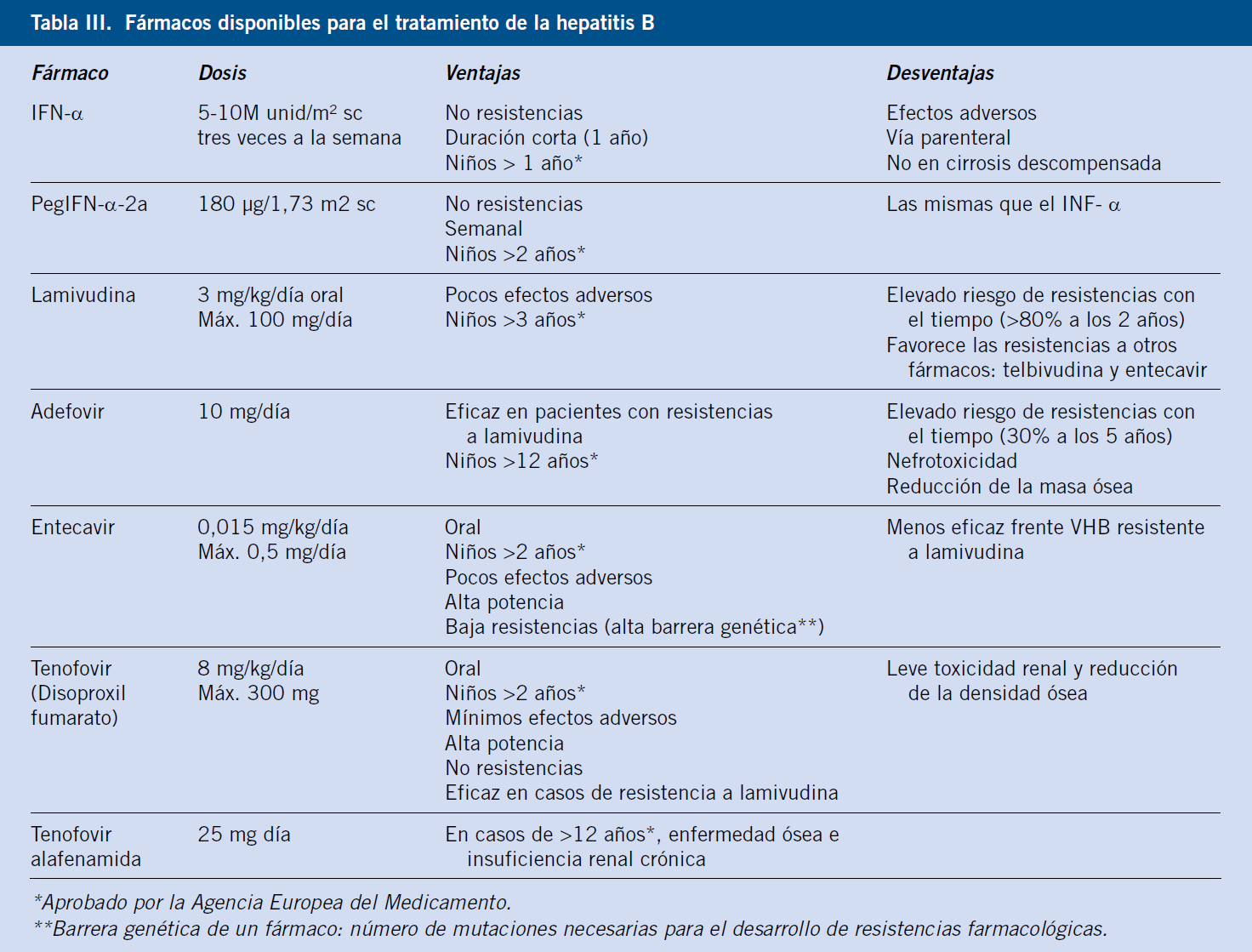
El periodo de latencia entre la ingestión del tóxico y la aparición de los síntomas es muy variable, aunque se aconseja revisar retrospectivamente los 3 meses previos. Una latencia inferior a 1 semana, así como erupción cutánea, eosinofilia o afectación de otros órganos, es compatible con una reacción de hipersensibilidad. Ocasionalmente, la sintomatología puede aparecer incluso semanas después de suspender el tratamiento, como ocurre con la administración de amoxicilina-clavulánico(16). Aproximadamente un 11% de pacientes tratados con ácido valproico desarrollan elevación de aminotransferasas, sobre todo niños, pudiendo presentar enfermedad hepática en los 3-4 primeros meses de iniciado el tratamiento(16). Podemos ver los principales fármacos productores de toxicidad hepática en la tabla III.

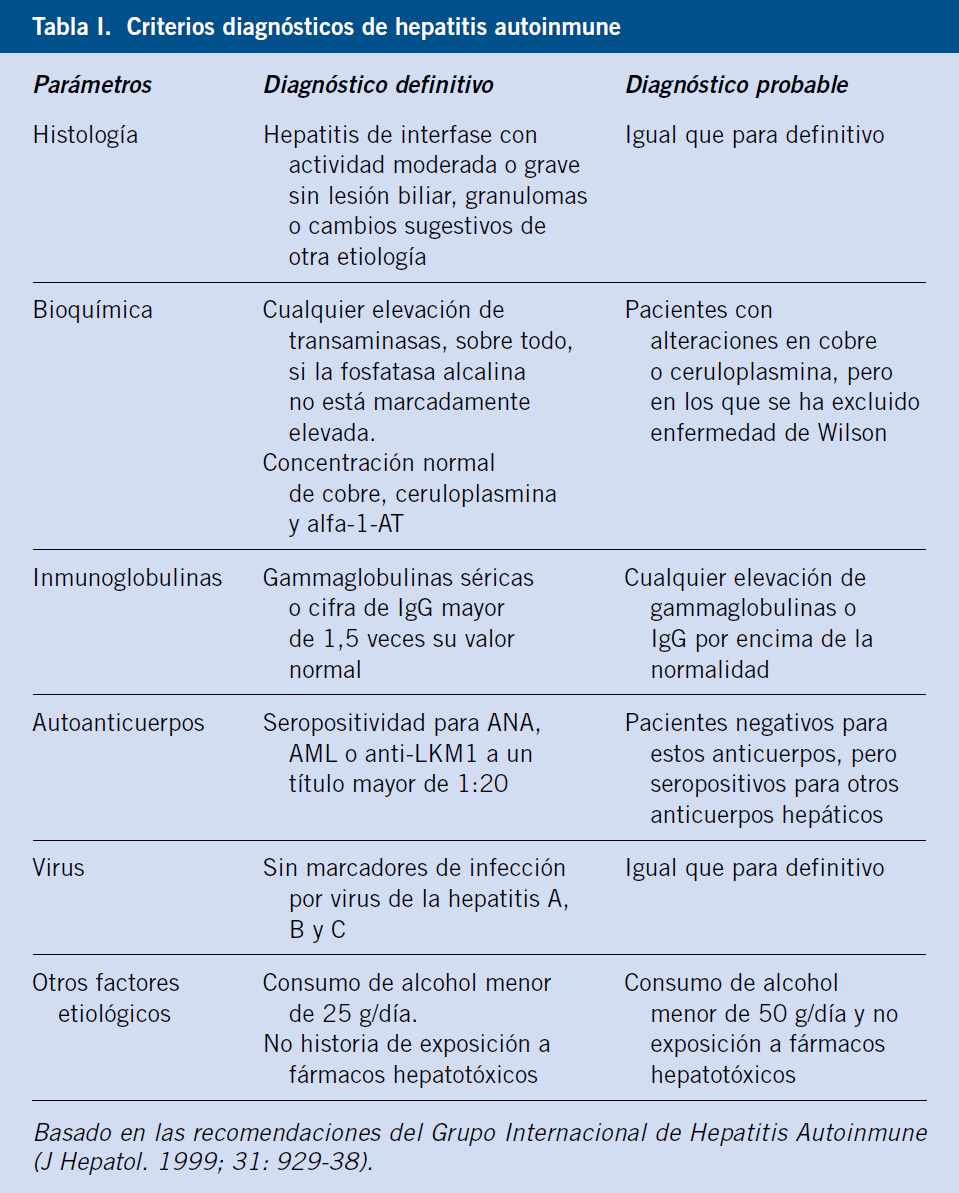
• Hepatitis autoinmune. Proceso que puede pasar desapercibido o debutar con síntomas de hepatitis aguda. Su diagnóstico precoz es prioritario, ya que sin tratamiento evoluciona hacia cirrosis e insuficiencia hepática. Se clasifica de acuerdo a su perfil serológico en: tipo 1 (60% de los casos en niños), con positividad para anticuerpos antinucleares (ANA), antimúsculo liso (SMA) o ambos, o con menos frecuencia, anti-antígeno soluble hepático (SLA), que afecta tanto a niños como adultos; y tipo 2, con positividad para anticuerpos antimicrosomas de hígado-riñón (LKM1) y/o anticitosol hepático tipo 1 (LC-1), fundamentalmente pediátrica(5,16).
Suele existir una importante elevación de transaminasas, sin elevación marcada de la fosfatasa alcalina ni de la GGT. La concentración de gamma- globulinas séricas o IgG total está elevada en más del 80% de los casos, y debe ser mayor de 1,5 veces su límite superior para valorarla en los scores diagnósticos.
La colangitis esclerosante primaria (CEP), probablemente también de origen inmune y asociada frecuentemente a una enfermedad inflamatoria intestinal, suele manifestarse con clínica de colestasis. Puede tener positivos los anticuerpos pANCA (anticitoplasma de neutrófilos) y puede evolucionar a una cirrosis biliar(16,17).
• Deficiencia de alfa-1-antitripsina (A1A). Es la causa más frecuente de enfermedad hepática crónica de base genética. Los fenotipos patológicos claramente relacionados con niveles más bajos de A1A y hepatopatía son el PiZZ y PiSZ(18). La mayoría de niños PiZZ se encuentran asintomáticos, pero un 10-15% desarrolla enfermedad hepática y evolución a cirrosis, y una parte requerirá un trasplante hepático, incluso en la edad pediátrica. La afectación hepática puede manifestarse precozmente, como colestasis neonatal o enfermedad hemorrágica aguda del lactante, o bien como hepatitis crónica, cirrosis e hipertensión portal en el niño mayor o adulto, pero muchas veces es detectada a raíz del hallazgo casual de hipertransaminasemia en un paciente asintomático.
Hay que tener en cuenta que la A1A es un reactante de fase aguda y que su concentración puede estar incrementada en respuesta a una inflamación, lo que produciría un resultado falso negativo. El diagnóstico definitivo se establece mediante la determinación fenotípica, o preferiblemente, del genotipo de la misma(5). Puede precisar estudio evolutivo con biopsia hepática.
• Enfermedad de Wilson. No hay ninguna prueba de laboratorio tan sensible y específica que permita por sí sola el diagnóstico de enfermedad de Wilson, aunque el hallazgo de una ceruloplasmina sérica inferior a 20 mg/dL, en un individuo joven con una alteración de las transaminasas, debe orientar al mismo. La ceruloplasmina es un reactante de fase aguda que puede estar alterado en diferentes circunstancias (malnutrición, proceso infeccioso o inflamatorio), existiendo un 5-15% de los pacientes con niveles normales (falsos negativos). También, sugerirán este diagnóstico la existencia del anillo de Kayser-Fleischer corneal y la presencia de una excreción urinaria de cobre en orina de 24 horas superior a 100 mg/día, y en caso de dudas, podemos llevar a cabo un test de sobrecarga previo con penicilamina. El diagnóstico se confirma generalmente demostrando la elevada concentración de cobre en tejido obtenido por biopsia hepática(16,19).
• Hemocromatosis. Debe sospecharse ante un índice de saturación de la transferrina IST > 45%, y ferritina aumentada, y puede confirmarse con el estudio genético en caso de obtener un IST elevado en, al menos, dos determinaciones. Es autosómica recesiva y el gen de la hemocromatosis (HFE) está situado en el brazo corto del cromosoma 6.
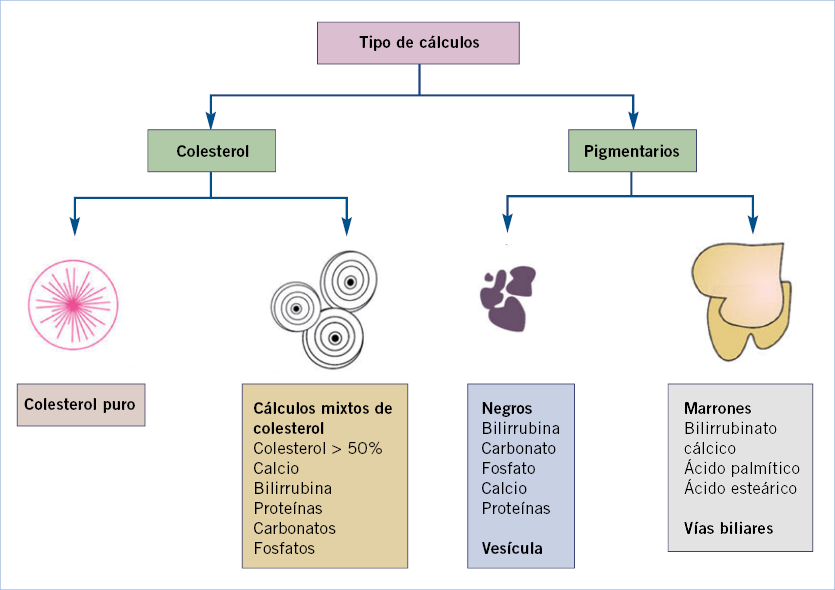
• Enfermedades hepatobiliares. Las principales son: las colestasis intrahepáticas familiares, atresia biliar extrahepática, quiste de colédoco, síndrome de Alagille, etc.
• Enfermedades metabólicas. Entre otras: galactosemia, tirosinemia, intolerancia hereditaria a la fructosa, glucogenosis, déficit de ornitina trascarbamilasa, porfirias o enfermedades de depósito (Gaucher, Niemann-Pick). Alguna podría detectarse en despistaje neonatal. Puede precisar realización de aminoácidos y ácidos orgánicos, cuerpos reductores en orina, o estudios enzimáticos y genéticos.
• Enfermedad celiaca. El 50% de los casos presentan una hipertransaminasemia leve al diagnóstico, que se normaliza tras la exclusión de gluten(20). Estaría implicada hasta en un 10% de casos de aumento de aminotransferasas no explicados por otras causas(9).
• Enfermedades musculares. Entre ellas destacan las distrofias musculares. En estos casos, se produce elevación de transaminasas, pero, sobre todo, de aldolasa y, más específica aún de miopatía, de creatinfosfoquinasa, así como la aparición de mioglobinuria.
• Miscelánea. Otras muchas enfermedades pueden alterar las pruebas hepáticas: enfermedades sistémicas con afectación del hígado (infecciones, hemopatías malignas, conectivopatías y enfermedades reumáticas, amiloidosis, insuficiencia cardiaca congestiva), insuficiencia pancreática (síndrome de Shwachman, fibrosis quística), nutrición parenteral prolongada, hipobetalipoproteinemia, intolerancia a proteínas vacunas, neoplasias primarias o metastásicas, síndrome de Budd-Chiari, etc. Se ha descrito aumento de transaminasas, tanto en el hipertiroidismo (hasta en un 27-37% de los casos)(2) como en el hipotiroidismo, o incluso signos de colestasis, sobre todo, en el lactante.
• Macro-GOT. Macroenzima o complejo de alto peso molecular formado por GOT y otros componentes del plasma. En niños, a diferencia del adulto, suele ser un proceso benigno y puede explicar hasta un tercio de casos con elevación exclusiva de GOT. Puede sospecharse midiendo el porcentaje de GOT precipitada con polietilenglicol, y el diagnóstico se confirma con electroforesis.
Actitud ante sospecha de hepatopatía
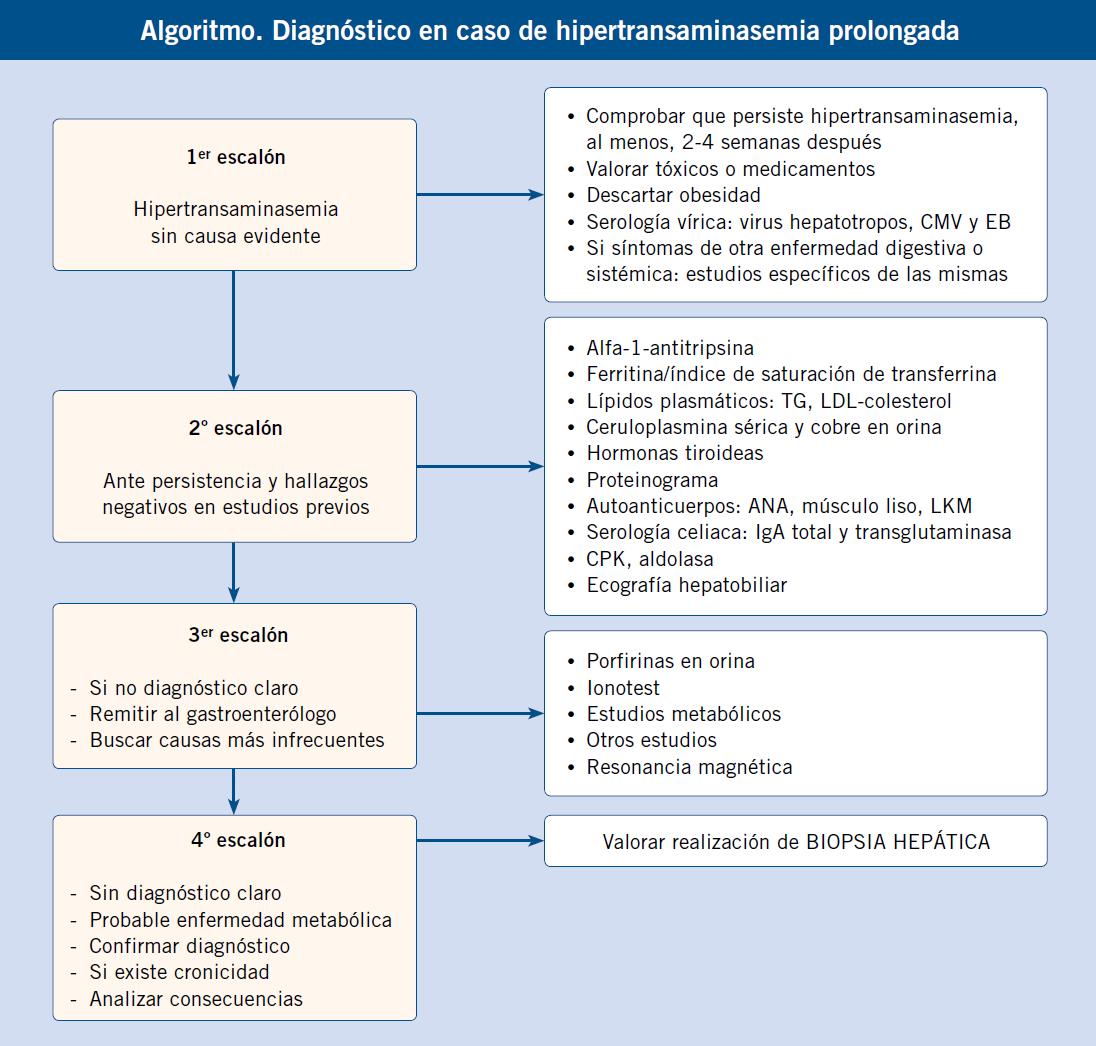
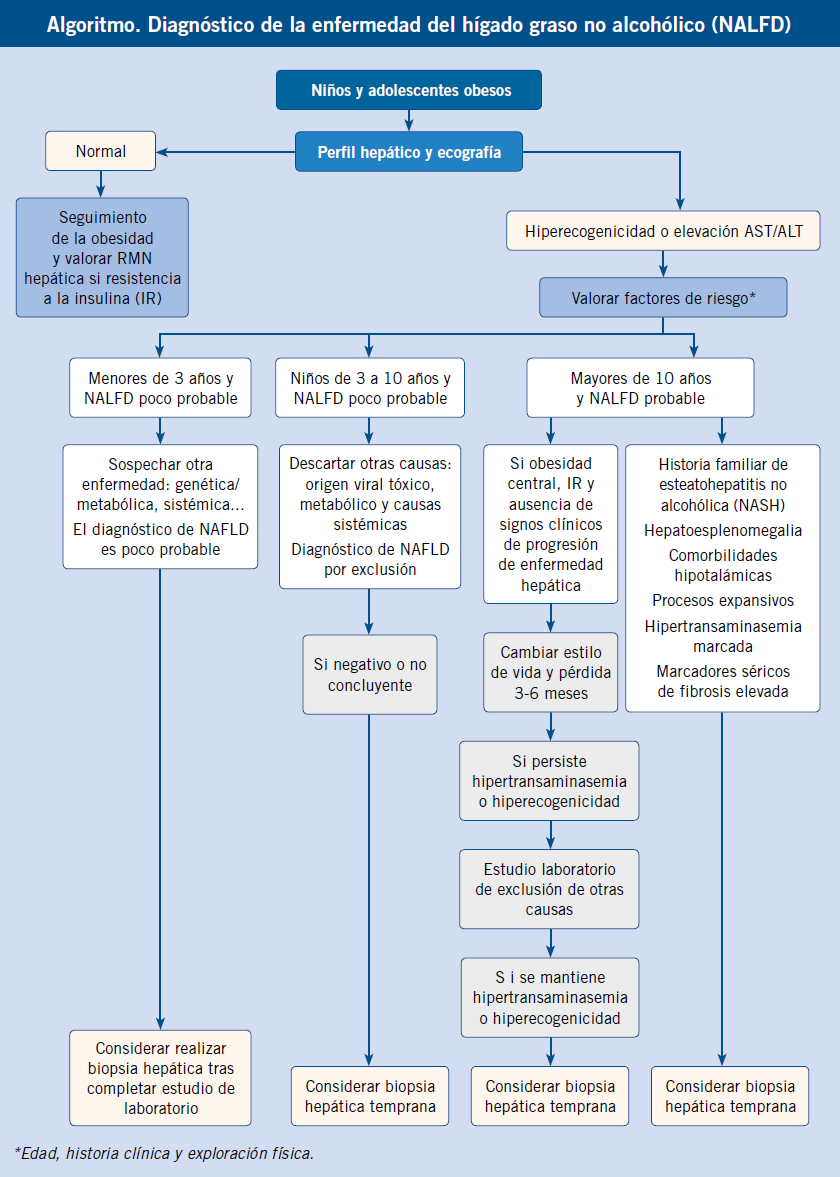
Debemos descartar una hiperbilirrubinemia conjugada ante toda ictericia prolongada más allá de las dos semanas de vida. El diagnóstico de una hipertransaminasemia mantenida requiere una actitud diagnóstica escalonada, como se describe a continuación.
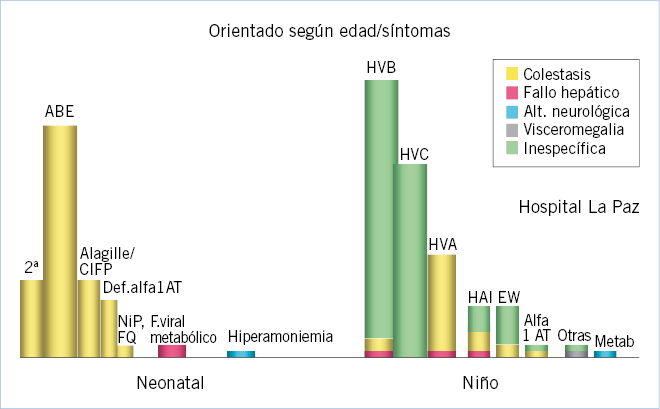
A partir del periodo neonatal, momento en el que por su inmadurez toda enfermedad grave suele asociar manifestaciones hepáticas, las principales causas de enfermedad hepática grave en lactantes son de tipo predominantemente colestásico, como: atresia biliar extrahepática, colestasis intrahepáticas familiares, síndrome de Alagille, metabolopatías y déficit de alfa-1-antitripsina(4).
Debemos detectarlas precozmente durante el periodo neonatal o la lactancia, por lo que es obligatorio realizar una determinación de bilirrubina conjugada a todos los neonatos que siguen ictéricos a los 15 días de edad.
Fuera del periodo neonatal y la lactancia, las causas son más variadas y se manifiestan ya más raramente con síntomas colestásicos. La hepatopatía suele detectarse de una manera casual a partir de síntomas inespecíficos, por alteraciones analíticas en exploraciones por enfermedades infecciosas intercurrentes o en controles de salud rutinarios(4).
Un hecho clínico muy frecuente, por tanto, es el hallazgo de una hipertransaminasemia, muchas veces de origen no aclarado, que supone un reto en el diagnóstico y seguimiento para el pediatra de Atención Primaria, dada la enorme variedad de causas que pueden ocasionarla. Intentaremos resumir en el algoritmo, la actitud a seguir en estos casos.
Elevaciones de las transaminasas inferiores a 2 veces el límite alto de la normalidad, deben ser comprobadas, al menos, dos semanas después (incluso cuatro, según la situación clínica), antes de iniciar cualquier tipo de estudio, ya que lo más probable es que hayan vuelto a la normalidad. En caso de hipertransaminasemia mantenida y prolongada, deberemos realizar estudios escalonadamente(9) (Algoritmo).
Bibliografía
Los asteriscos muestran el interés del artículo a juicio del autor.
1.** González Jiménez D, Santos Rodríguez PM. Hipertransaminasemia en Pediatría. Bol pediatr. 2013; 53: 137-45.
2.** Martínez-Valverde A, Lastra G. Exploración del hígado. En: Galdó A, Cruz M (eds.), Tratado de Exploración clínica en Pediatría. Masson. Barcelona, 1995; 429-45.
3. Muñoz Bartolo G. Hepatomegalia. Pediatr Integral. 2015; XIX(3): 180-97.
4.** de la Vega A, Frauca Remacha E. Síndrome colestático. Actitud diagnóstico-terapéutica. Pediatr Integral. 2015; XIX(3): 168-79.
5.*** Squires JE, Balistreri WF. Manifestaciones de las enfermedades hepáticas. Evaluación de los pacientes con sospecha de enfermedad hepática. En: Kliegman RM, et al (eds), Nelson Tratado de Pediatría 20ª ed. Elsevier. 2016; 2: 2012-8.
6. Fraser A, Longnecker M, Lawlor D. Prevalence of elevated alanine aminotransferasa among US adolescents and associated factors: NHANES 1999-2004. Gastroenterology. 2007; 133: 1814-20.
7.*** Maldonado Lozano J. Valoración de la función hepática. En: Argüelles Martín F y cols. (eds.). Tratado de Gastroenterología, hepatología y nutrición pediátrica aplicada de la SEGHNP. Ergon; 2011. p. 419-26.
8.** Codoceo Alquinta R, Perdomo Giraldi M, Muñoz Codoceo R. Interpretación del laboratorio en gastroenterología: Estudio de la función hepática. En: SEGHNP (ed), Tratamiento en gastroenterología, hepatología y nutrición pediátrica, 3ª ed. Ergon; 2012. p. 360-1.
9.*** García Martín M, Zurita Molina A. Transaminasas: Valoración y significación clínica. Protocolos de la AEP. Protocolos de Gastroenterología, Hepatología y Nutrición. Asociación Española de Pediatría, Sociedad Española de Gastroenterología, Hepatología y Nutrición Pediátrica, 2ª ed. Ergon; 201. p. 267-75.
10. Bugeac N, Pacht A, Mandel H, Iancu T, Tamir A, Srugo I, et al. The significance of isolated elevation of serum aminotransferases in infants and young children. Arch Dis Child. 2007; 92: 1109-12.
11. Tripodi A, Caldwell SH, Hoffman M, Trotter JF, Sanyal AJ. Review article: the prothrombin time test as a measure of bleeding risk and prognosis in liver disease. Aliment Pharmacol Ther. 2007; 15: 141-8.
12. Picó Aliaga SD, Muro Velilla D, García-Martí G, Sangüesa Nebot C, Martí-Bonmatí L. Acoustic radiation force impulse imaging elastography is efficacious in detecting hepatic fibrosis in children. Radiologia. 2015; 57: 314-20.
13.** Marugán de Miguelsanz JM, Torres Hinojal MC. Hepatitis agudas. Pediatr Integral. 2015; XIX(3): 222-8.
14. González de Caldas Marchal R, Millán Jiménez A, Blanco Rodríguez M. Hígado graso en la infancia. En: SEGHNP (ed), Tratamiento en Gastroenterología, Hepatología y Nutrición pediátrica, 4ª ed. Ergon; 2016. p. 509-21.
15. Zurita Molina A, de la Rubia Fernández L. Hepatitis medicamentosa y tóxica. En: SEGHNP (ed), Tratamiento en gastroenterología, hepatología y nutrición pediátrica, 4ª ed. Ergon; 2016. p. 459-68.
16. Urruzuno Tellería P, Camarena Grande C. Hepatopatías autoinmunes. En: SEGHNP (ed), Tratamiento en Gastroenterología, Hepatología y Nutrición pediátrica, 4ª ed. Ergon; 2016. p. 441-58.
17. Sokollik C, McLin VA, Vergani D, Terziroli Beretta-Piccoli B, Mieli-Vergani G. Juvenile autoimmune hepatitis: A comprehensive review. J Autoimmun. 2018; 95: 69-76.
18. Legarda Tamará M, Balmaseda Serrano EM, Gutiérrez Junquera C. Déficit de alfa-1-antitripsina. En: SEGHNP (ed), Tratamiento en Gastroenterología, Hepatología y Nutrición pediátrica, 4ª ed. Ergon; 2016. p. 499-509.
19. Peña Quintana L, de la Vega Bueno A. Enfermedad de Wilson. En: SEGHNP (ed), Tratamiento en Gastroenterología, Hepatología y Nutrición pediátrica, 4ª ed. Ergon; 2016. p. 485-99.
20. Kwiatek-?redzi?ska KA, Kondej-Muszy?ska K, U?cinowicz M, Werpachowska I, Sobaniec-?otowska M, Lebensztejn D. Liver pathology in children with newly diagnosed celiac disease. Clin Exp Hepatol. 2019; 5: 129-32.
Bibliografía recomendada
- Martínez-Valverde A, Lastra G. Exploración del hígado. En: Galdó A, Cruz M (eds.), Tratado de Exploración clínica en Pediatría. Masson. Barcelona, 1995; 429-45.
Antigua, pero clásica revisión de las principales manifestaciones relacionadas con las enfermedades hepáticas, centrada, sobre todo, en la semiología clínica.
- Squires JE, Balistreri WF. Manifestaciones de las enfermedades hepáticas. Evaluación de los pacientes con sospecha de enfermedad hepática. En: RM Kliegman, et al (eds), Nelson Tratado de Pediatría 20ª ed. Elsevier. 2016; 2: 2012-8.
Extenso y completo capítulo dedicado a la evaluación de pacientes con sospecha de enfermedad hepática, en el Tratado de Pediatría con más referencias en el mundo.
- Maldonado Lozano J. Valoración de la función hepática. En: Argüelles Martín F y cols. (eds.), Tratado de Gastroenterología, hepatología y nutrición pediátrica aplicada de la SEGHNP. Ergon; 2011. p. 419-26.
Amplio capítulo dedicado a las pruebas de función hepática, en el primer tratado de Gastroenterología, Hepatología y Nutrición pediátrica publicado en español.
- Codoceo Alquinta R, Perdomo Giraldi M, Muñoz Codoceo R. Interpretación del laboratorio en gastroenterología: Estudio de la función hepática. En: SEGHNP (ed), Tratamiento en gastroenterología, hepatología y nutrición pediátrica, 3ª ed. Ergon; 2012. p. 360-1.
Interesante revisión de las pruebas de función hepática, útil para la interpretación de sus resultados, en un manual muy práctico de diagnóstico y tratamiento de las patologías digestivas más frecuentes, publicado por la Sociedad Española de Gastroenterología, Hepatología y Nutrición pediátricas.
- García Martín M, Zurita Molina A. Transaminasas: Valoración y significación clínica. Protocolos de la AEP. Protocolos de Gastroenterología, Hepatología y Nutrición. Asociación Española de Pediatría, Sociedad Española de Gastroenterología, Hepatología y Nutrición Pediátrica 2010, 2ª ed. Ergon; 2010. p. 267-75.
Breve y concisa revisión sobre el significado y la actitud a seguir ante una hipertransaminasemia crónica en la práctica clínica. Forma parte de los protocolos auspiciados por la Sociedad Española de Gastroenterología, Hepatología y Nutrición pediátricas, y la Asociación Española de Pediatría.
| Caso clínico |
|
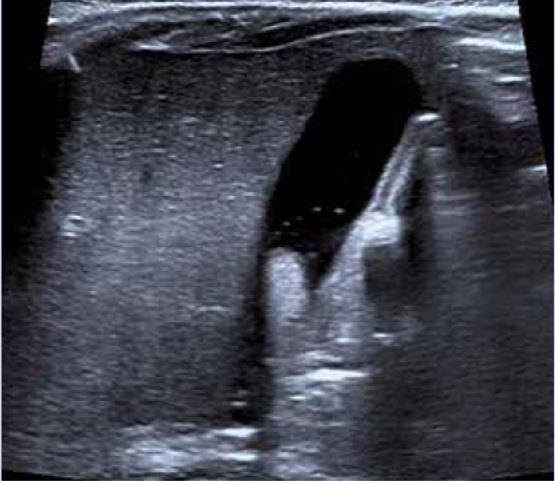
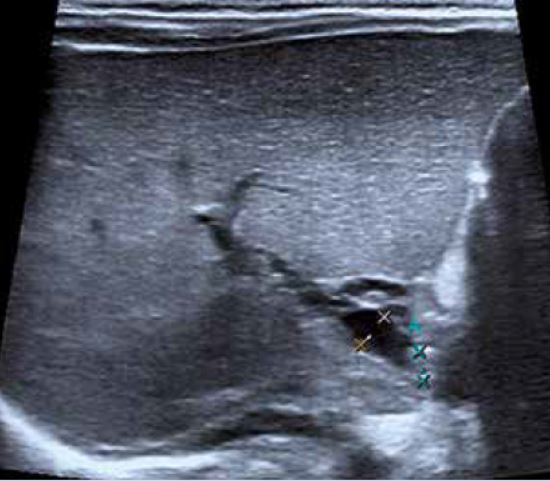
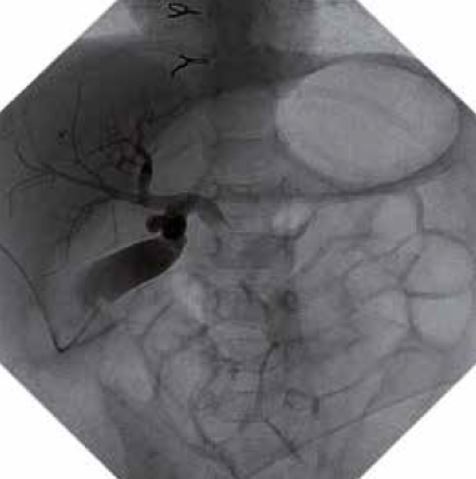
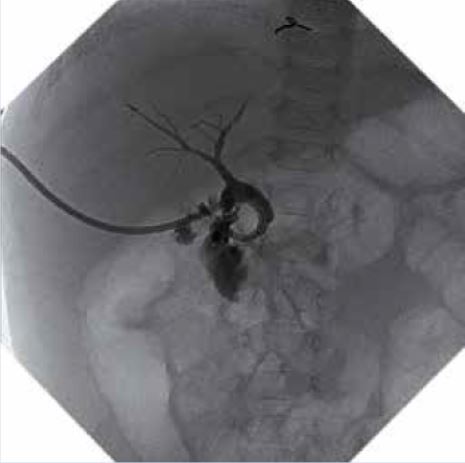
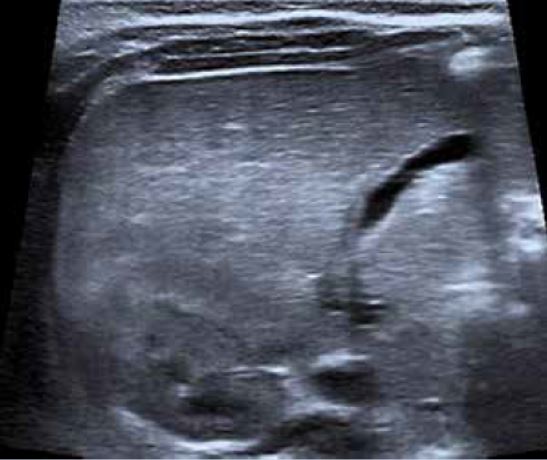
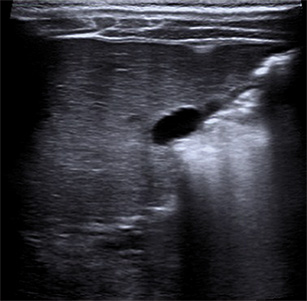
Varón de 5 años de edad que consulta por hipertransaminasemia detectada en analítica realizada por su pediatra. Entre los antecedentes familiares, no hay nada de interés. Es el primer hijo, fruto de un embarazo y parto normales, nacido pretérmino a las 36 semanas con un peso de 2.450 g y 46 cm de longitud. Presenta una ictericia neonatal catalogada de ictericia fisiológica prolongada, que no precisó fototerapia. Lactancia materna exclusiva, con buen apetito. A los 10 días, habiendo recuperado el peso al nacimiento, su pediatra comprueba la persistencia de la ictericia, realizando una analítica, que muestra un hemograma y transaminasas normales, y BRT de 8 mg/dl, con una BRI de 4,5 mg/dl, y BRD 3,5 mg/dl. El resto de la exploración física es normal, y no ha presentado coluria ni acolia. La ecografía hepatobiliar es normal, y se realiza un HIDA-Tc, con presencia de trazador a nivel intestinal. La ictericia desaparece en el segundo mes de vida. Su crecimiento y desarrollo posterior es normal, sin otras patologías de interés, excepto: una bronquiolitis que no precisó ingreso, varias infecciones respiratorias de vías altas y dos episodios de diarrea, el último de los cuales se prolonga en el tiempo, con estacionamiento ponderal. Por este motivo, su médico realiza una analítica ordinaria, con: hemograma, bioquímica, coprocultivo y virus en heces, y serología de celiaca, toda ella normal, excepto una GOT de 65 y GPT de 80 U/L. La repetición de la misma 8 días después, permite comprobar la persistencia de esta discreta hipertransaminasemia, por lo que es remitido a nuestra consulta, ante la sospecha de hepatitis crónica. En nuestra consulta, se repite el mismo estudio y se amplían las técnicas diagnósticas, lo que permite llegar al diagnóstico del proceso.
|